
REFRAME YOUR APPROACH WITH THE TUKYSA REGIMEN*
TUKYSA is the first FDA-approved HER2-directed therapy in 2L+ RAS WT, HER2+ mCRC1,2
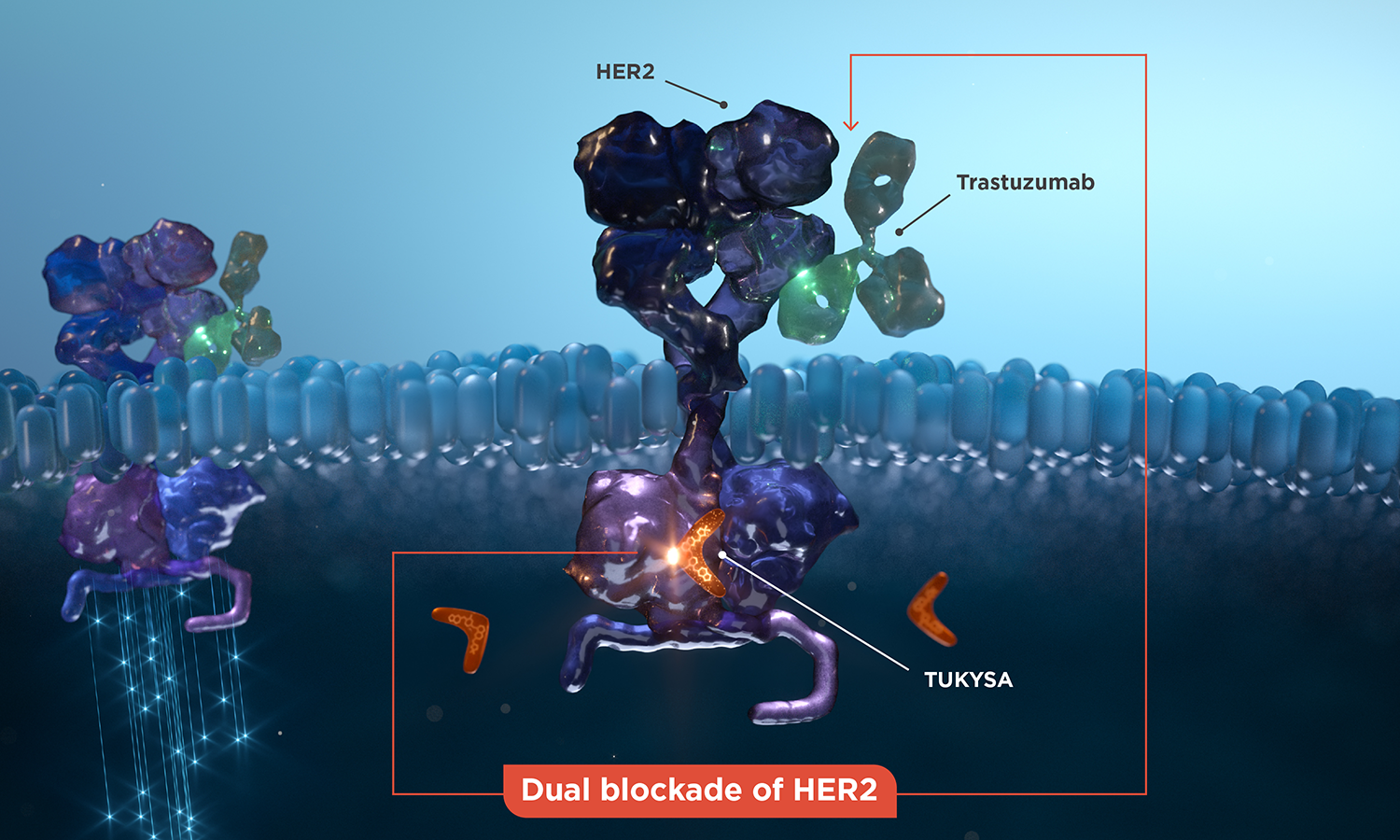
Proposed mechanism of action
TUKYSA is part of a chemotherapy-free treatment option designed
to target HER2+ mCRC1,5*
Dual blockade of HER2
The TUKYSA intracellular blockade of HER2 is thought to complement the trastuzumab blockade on the extracellular HER2 domain. The combination has demonstrated an increased antitumor effect compared with either product alone in animal models.1,6
Keep current on TUKYSA® (tucatinib). Register for email updates.
Important Safety Information
Warnings and Precautions
- Diarrhea: TUKYSA can cause severe diarrhea including dehydration, hypotension, acute kidney injury, and death. If diarrhea occurs, administer antidiarrheal treatment as clinically indicated. Perform diagnostic tests as clinically indicated to exclude other causes of diarrhea. Based on the severity of the diarrhea, interrupt dose, then dose reduce or permanently discontinue TUKYSA. In MOUNTAINEER, when TUKYSA was given in combination with trastuzumab, diarrhea occurred in 64% of patients, including Grade 3 (3.5%), Grade 2 (10%), and Grade 1 (50%).
- Hepatotoxicity: TUKYSA can cause severe hepatotoxicity. Monitor ALT, AST, and bilirubin prior to starting TUKYSA, every 3 weeks during treatment, and as clinically indicated. Based on the severity of hepatotoxicity, interrupt dose, then dose reduce or permanently discontinue TUKYSA. In MOUNTAINEER, 6% of patients had a bilirubin increase > 3 × ULN (Grade ≥3), 6% had an AST increase > 5 × ULN, and 4.7% had an ALT increase > 5 × ULN. Hepatotoxicity led to dose reduction of TUKYSA in 3.5% of patients and discontinuation of TUKYSA in 2.3% of patients.
- Embryo-Fetal Toxicity: TUKYSA can cause fetal harm. Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females of reproductive potential, and male patients with female partners of reproductive potential, to use effective contraception during TUKYSA treatment and for 1 week after the last dose.
Adverse Reactions
Serious adverse reactions occurred in 22% of patients; the most common (in ≥2% of patients) were intestinal obstruction (7%), urinary tract infection (3.5%), pneumonia, abdominal pain, and rectal perforation (2.3% each).
Adverse reactions leading to permanent discontinuation of TUKYSA occurred in 6% of patients; the most common (in ≥2% of patients) was increased ALT (2.3%). Adverse reactions leading to dosage interruption occurred in 23% of patients; the most common (in ≥3% of patients) were increased ALT and diarrhea (3.5% each). Adverse reactions leading to dose reduction occurred in 9% of patients; the most common (in ≥2% of patients) were increased ALT and diarrhea (2.3% each).
The most common adverse reactions (≥20%) in patients treated with TUKYSA and trastuzumab were diarrhea, fatigue, rash, nausea, abdominal pain, infusion-related reactions, and pyrexia.
Other adverse reactions (<10%) include epistaxis (7%), weight decreased (7%), oropharyngeal pain (5%), oral dysesthesia (1%), and stomatitis (1%).
Lab Abnormalities
In MOUNTAINEER, Grade ≥3 laboratory abnormalities reported in ≥5% of patients who received TUKYSA were decreased lymphocytes, decreased sodium, increased AST, and increased bilirubin.
The mean increase in serum creatinine was 32% within the first 21 days of treatment with TUKYSA. The serum creatinine increases persisted throughout treatment and were reversible in 87% of patients with values outside normal lab limits upon treatment completion. Consider alternative markers of renal function if persistent elevations in serum creatinine are observed.
Drug Interactions
- Strong CYP3A/Moderate CYP2C8 Inducers: Concomitant use may decrease TUKYSA activity. Avoid concomitant use of TUKYSA.
- Strong or Moderate CYP2C8 Inhibitors: Concomitant use of TUKYSA with a strong CYP2C8 inhibitor may increase the risk of TUKYSA toxicity; avoid concomitant use. Increase monitoring for TUKYSA toxicity with moderate CYP2C8 inhibitors.
- CYP3A Substrates: Concomitant use may increase the toxicity associated with a CYP3A substrate. Avoid concomitant use of TUKYSA with a CYP3A substrate, where minimal concentration changes may lead to serious or life-threatening toxicities. If concomitant use is unavoidable, decrease the CYP3A substrate dosage.
- P-gp Substrates: Concomitant use may increase the toxicity associated with a P-gp substrate. Consider reducing the dosage of P-gp substrates, where minimal concentration changes may lead to serious or life-threatening toxicities.
Use in Specific Populations
- Lactation: Advise women not to breastfeed while taking TUKYSA and for 1 week after the last dose.
- Hepatic Impairment: Reduce the dose of TUKYSA for patients with severe (Child-Pugh C) hepatic impairment.
REF-T1K1163
Indication
TUKYSA is indicated in combination with trastuzumab for the treatment
of adult patients with RAS wild-type, HER2-positive
unresectable or metastatic colorectal cancer that has progressed
following treatment with fluoropyrimidine-, oxaliplatin-, and
irinotecan-based chemotherapy.
This indication is approved under accelerated approval based on tumor
response rate and durability of response. Continued approval for this
indication may be contingent upon verification and description of
clinical benefit in confirmatory trials.
Please see full Prescribing Information.
Important Safety Information
Important Safety Information and Indication
Indication
Warnings and Precautions
Warnings and Precautions
-
Diarrhea:
TUKYSA can cause severe diarrhea including dehydration,
hypotension, acute kidney injury, and death. If diarrhea occurs,
administer antidiarrheal treatment as clinically indicated.
Perform diagnostic tests as clinically indicated to exclude
other causes of diarrhea. Based on the severity of the diarrhea,
interrupt dose, then dose reduce or permanently discontinue
TUKYSA.
In MOUNTAINEER, when TUKYSA was given in combination with trastuzumab, diarrhea occurred in 64% of patients, including Grade 3 (3.5%), Grade 2 (10%), and Grade 1 (50%). - Hepatotoxicity: TUKYSA can cause severe hepatotoxicity. Monitor ALT, AST, and bilirubin prior to starting TUKYSA, every 3 weeks during treatment, and as clinically indicated. Based on the severity of hepatotoxicity, interrupt dose, then dose reduce or permanently discontinue TUKYSA. In MOUNTAINEER, 6% of patients had a bilirubin increase > 3 × ULN (Grade ≥3), 6% had an AST increase > 5 × ULN, and 4.7% had an ALT increase > 5 × ULN. Hepatotoxicity led to dose reduction of TUKYSA in 3.5% of patients and discontinuation of TUKYSA in 2.3% of patients.
-
Embryo-Fetal Toxicity:
TUKYSA can cause fetal harm. Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females of reproductive potential, and male patients with female partners of reproductive potential, to use effective contraception during TUKYSA treatment and for 1 week after the last dose.
Adverse Reactions
Serious adverse reactions occurred in 22% of patients; the most common (in ≥2% of patients) were intestinal obstruction (7%), urinary tract infection (3.5%), pneumonia, abdominal pain, and rectal perforation (2.3% each).
Adverse reactions leading to permanent discontinuation of TUKYSA occurred in 6% of patients; the most common (in ≥2% of patients) was increased ALT (2.3%). Adverse reactions leading to dosage interruption occurred in 23% of patients; the most common (in ≥3% of patients) were increased ALT and diarrhea (3.5% each). Adverse reactions leading to dose reduction occurred in 9% of patients; the most common (in ≥2% of patients) were increased ALT and diarrhea (2.3% each).
The most common adverse reactions (≥20%) in patients treated with TUKYSA and trastuzumab were diarrhea, fatigue, rash, nausea, abdominal pain, infusion-related reactions, and pyrexia.
Other adverse reactions (<10%) include epistaxis (7%), weight decreased (7%), oropharyngeal pain (5%), oral dysesthesia (1%), and stomatitis (1%).
Lab Abnormalities
In MOUNTAINEER, Grade ≥3 laboratory abnormalities reported in ≥5% of patients who received TUKYSA were decreased lymphocytes, decreased sodium, increased AST, and increased bilirubin.
The mean increase in serum creatinine was 32% within the first 21 days of treatment with TUKYSA. The serum creatinine increases persisted throughout treatment and were reversible in 87% of patients with values outside normal lab limits upon treatment completion. Consider alternative markers of renal function if persistent elevations in serum creatinine are observed.
Drug Interactions
- Strong CYP3A/Moderate CYP2C8 Inducers: Concomitant use may decrease TUKYSA activity. Avoid concomitant use of TUKYSA.
- Strong or Moderate CYP2C8 Inhibitors: Concomitant use of TUKYSA with a strong CYP2C8 inhibitor may increase the risk of TUKYSA toxicity; avoid concomitant use. Increase monitoring for TUKYSA toxicity with moderate CYP2C8 inhibitors.
- CYP3A Substrates: Concomitant use may increase the toxicity associated with a CYP3A substrate. Avoid concomitant use of TUKYSA with a CYP3A substrate, where minimal concentration changes may lead to serious or life-threatening toxicities. If concomitant use is unavoidable, decrease the CYP3A substrate dosage.
- P-gp Substrates: Concomitant use may increase the toxicity associated with a P-gp substrate. Consider reducing the dosage of P-gp substrates, where minimal concentration changes may lead to serious or life-threatening toxicities.
Use in Specific Populations
- Lactation: Advise women not to breastfeed while taking TUKYSA and for 1 week after the last dose.
- Hepatic Impairment: Reduce the dose of TUKYSA for patients with severe (Child-Pugh C) hepatic impairment.
REF-T1K1163
Indication
TUKYSA is indicated in combination with trastuzumab for the
treatment of adult patients with RAS wild-type,
HER2-positive unresectable or metastatic colorectal cancer that
has progressed following treatment with fluoropyrimidine-,
oxaliplatin-, and irinotecan-based chemotherapy.
This indication is approved under accelerated approval based on
tumor response rate and durability of response. Continued approval
for this indication may be contingent upon verification and
description of clinical benefit in confirmatory trials.
Please see full Prescribing Information.
TUKYSA is indicated in combination with trastuzumab for the treatment
of adult patients with RAS wild-type, HER2-positive
unresectable or metastatic colorectal cancer that has progressed
following treatment with fluoropyrimidine-, oxaliplatin-, and
irinotecan-based chemotherapy.
This indication is approved under accelerated approval based on tumor
response rate and durability of response. Continued approval for this
indication may be contingent upon verification and description of
clinical benefit in confirmatory trials.
Please see full Prescribing Information.